Basics of Cushing’s Syndrome



Cushing’s syndrome is a rare disease resulting from chronic exposure to excessive circulating levels of cortisol produced from tumours in the pituitary or adrenal glands or from other sources.
This introductory section on Cushing’s Hub is designed for non-specialists who may be largely unfamiliar with the disorder, such as medical students, GPs, or specialists from other fields. Here you will find an explanation of the symptoms of Cushing’s syndrome in adults together with a detailed description of the pathophysiology and its resultant impact on morbidity and mortality.
Harvey Cushing
In 1912, Harvey Cushing reported an endocrinological syndrome caused by a malfunction of the pituitary gland which he termed ‘polyglandular syndrome’, and which came to be known as Cushing’s syndrome. Cushing developed many foundational techniques and practices in neurosurgery, for example, the use of X-rays to diagnose brain tumours, the use of electrical stimuli to study the human sensory cortex, and the development of the Bovie electrocautery tool with William T. Bovie, a physicist.
Cushing was also the world’s leading teacher of neurosurgeons in the early 20th century and considerably improved the survival of patients after difficult brain operations for intracranial tumours.
Cushing’s Awareness Day is celebrated on April 8th, the birthday of Harvey Cushing.
The Disease Overview section details the history of Cushing’s syndrome; its prevalence; the HPA axis – interactions between the hypothalamus, pituitary and adrenal glands; and its wide range of presentations and symptoms, such as hypertension, rounded face, thin skin and osteoporosis. The Diagnosis section provides details of several diagnostic methods, such as the 24-hour urinary free cortisol test, late-night salivary cortisol test, overnight dexamethasone suppression test and desmopressin test, as well as information on differential diagnoses to help find the cause of the disease. Treatment goals, the different comorbidities, current guidelines, transsphenoidal surgery, radiotherapy, bilateral adrenalectomy, pharmacological interventions and future treatments can be found in the Disease Management section. The section concludes with a range of helpful Patient Resources.
Contents
Disease overview
Cushing’s Syndrome can be challenging to detect by family medicine or primary care practitioners due to its rarity and lack of specific symptoms. However, identifying symptoms of Cushing’s syndrome is vital for diagnosis and treatment to prevent irreversible organ damage as well as malignancies. Read on to learn more about the incidence, symptoms, and pathophysiology of this condition.
Cushing’s Syndrome
Cushing’s syndrome (CS) was first described by Harvey Cushing in 1932. It is a condition where the body produces excess cortisol over a long period of time, regardless of the physiological cause;1 therefore, it is often interchangeably called hypercortisolism. Cortisol is a steroid hormone produced by the adrenal glands and is sometimes called the “stress hormone” because it helps the body cope with stressors like illness or injury through its anti-inflammatory and immune-modulating properties. Most often, CS is caused by supraphysiological doses of exogenous glucocorticoids, such as prednisone, used for inflammation or corticosteroids used for asthma. Topical and injectable steroids may also cause the condition.1, 2
Epidemiology
CS is a rare disease with an incidence of 0.7–2.4 per million people per year.1, 3 However, its comorbidities are similar to those of more common disorders such as uncontrolled diabetes mellitus or hypertension, so the true incidence may be underestimated.3 CS is more common in women than men (ratio 3:1) and patients are typically aged ≥40 years at diagnosis.4, 5 The mortality rate is around four times higher in CS than in the general population, and even higher among women.3, 4
Pathophysiology
As its name suggests, the hypothalamic-pituitary-adrenal (HPA) axis involves a complex interplay between three glands: the hypothalamus, pituitary and adrenal glands (Figure 1). The HPA axis controls the synthesis of corticotrophin-releasing hormone in the hypothalamus, adrenocorticotrophic hormone (ACTH) in the anterior pituitary gland, and cortisol in the adrenal cortex. It functions mainly to produce basal cortisol rhythms and to respond to a wide variety of stimuli or stressors. A negative feedback system ensures that cortisol levels remain within a healthy physiological range.6
 Figure 1. The HPA axis.ACTH: adrenocorticotropic hormone; AVP: arginine vasopressin; CRH: corticotropin-releasing hormone.
Figure 1. The HPA axis.ACTH: adrenocorticotropic hormone; AVP: arginine vasopressin; CRH: corticotropin-releasing hormone.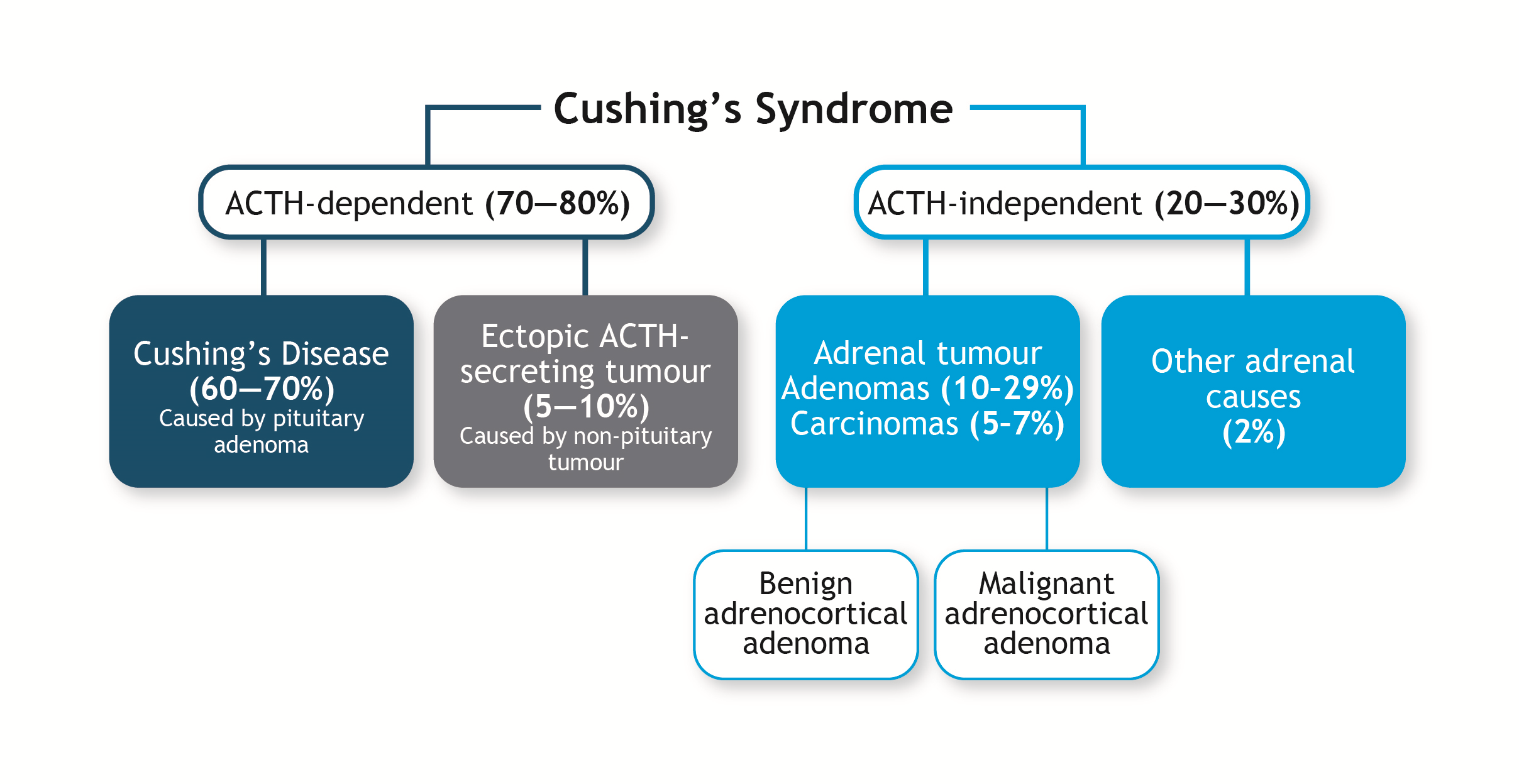 Figure 2. Classification of Cushing’s syndrome.5ACTH: adrenocorticotropic hormone.
Figure 2. Classification of Cushing’s syndrome.5ACTH: adrenocorticotropic hormone.Symptoms
Figure 3. Clinical characteristics of Cushing’s syndrome.1References
- Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet. 2006; 367: 1605-1617. https://pubmed.ncbi.nlm.nih.gov/16698415.
- Newell-Price J. Etiologies of Cushing’s syndrome. In: Bronstein MD, ed. Cushing’s Syndrome: Pathophysiology, Diagnosis and Treatment. Totowa, NJ: Humana Press, 2011; 21-29.
- Sharma ST, Nieman LK, Feelders RA. Cushing’s syndrome: epidemiology and developments in disease management. Clin Epidemiol 2015; 7: 281-293. https://pubmed.ncbi.nlm.nih.gov/25945066.
- Hakami OA, Ahmed S, Karavitaki N. Epidemiology and mortality of Cushing’s syndrome. Best Pract Res Clin Endocrinol Metab. 2021; 35: 101521. https://pubmed.ncbi.nlm.nih.gov/33766428.
- Lacroix A, Feelders RA, Stratakis CA, Nieman LK. Cushing’s syndrome. Lancet 2015; 386: 913-927. https://pubmed.ncbi.nlm.nih.gov/26004339.
- Raff H, Carroll T. Cushing’s syndrome: from physiological principles to diagnosis and clinical care. J Physiol. 2015; 593: 493-506. https://pubmed.ncbi.nlm.nih.gov/25480800.
- Barbot M, Zilio M, Scaroni C. Cushing’s syndrome: overview of clinical presentation, diagnostic tools and complications. Best Pract Res Clin Endocrinol Metab. 2020; 34: 101380. https://pubmed.ncbi.nlm.nih.gov/32165101.
- Stratakis CA. Cushing syndrome caused by adrenocortical tumors and hyperplasias (corticotropin- independent Cushing syndrome). Endocr Dev. 2008; 13: 117-132. https://pubmed.ncbi.nlm.nih.gov/18493137.
- Fassnacht M, Terzolo M, Allolio B, et al. Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med. 2012; 366: 2189-2197. https://pubmed.ncbi.nlm.nih.gov/22551107.
- Ioachimescu AG, Remer EM, Hamrahian AH. Adrenal incidentalomas: a disease of modern technology offering opportunities for improved patient care. Endocrinol Metab Clin North Am. 2015; 44: 335-354. https://pubmed.ncbi.nlm.nih.gov/26038204.
- Nieman LK, Biller BMK, Findling JW, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008; 93: 1526-1540. https://pubmed.ncbi.nlm.nih.gov/18334580.
- Bruno OD. Clinical features of Cushing’s syndrome. In: Bronstein MD, ed. Cushing’s Syndrome: Pathophysiology, Diagnosis and Treatment. Totowa, NJ: Humana Press, 2011; 53-64.
- Prague JK, May S, Whitelaw BC. Cushing’s syndrome. BMJ. 2013; 346: f945. https://pubmed.ncbi.nlm.nih.gov/23535464.
- Resmini E. Persistent comorbidities in Cushing’s syndrome after endocrine cure. Adv Endocrinol 2014; 2014: 231432.
Diagnosis
24-hour urinary free cortisol
Late-night salivary cortisol
Overnight 1-mg dexamethasone suppression test
The 1-mg DST takes advantage of the loss of glucocorticoid negative-feedback inhibition of corticotropin-releasing hormone (CRH) and adrenocorticotrophic hormone (ACTH) secretion in CS.6 Patients are given 1-mg of dexamethasone, a synthetic glucocorticoid, between 11:00 pm and midnight, and serum cortisol levels are measured between 8:00 am and 9:00 am the following morning. Normally dexamethasone suppresses ACTH and cortisol levels, but not in patients with CS. A serum cortisol reading of <1.8 µg/dL (50 nmol/L) excludes the diagnosis of CS with >95% sensitivity but 80% specificity.3 False-positive results are common, particularly in women on oral oestrogen therapy or patients taking anticonvulsants or medications that induce hepatic cytochrome P450 3A4 enzyme activity. Moreover, reduced clearance of dexamethasone, as seen in liver and renal failure, and decreased CBG concentrations in patients with concurrent nephrotic syndrome may lead to false-negative results. Therefore, some experts have advocated the simultaneous measurement of cortisol and dexamethasone for these tests to prevent false-positive and false-negative results and improve test interpretability.1, 3.6
Longer low-dose dexamethasone suppression test
In the longer low-dose DST, patients take dexamethasone in doses of 0.5 mg at 6-hour intervals for 48 hours. Serum cortisol is measured 6 hours after the last dose of dexamethasone. A final level of >1.81 µg/dL (50 nmol/L) is ~95% sensitive and specific for CS.2, 3 The longer low-dose DST is useful in patients for whom UFC measurements are less useful as an initial test, such as patients with psychiatric conditions, alcoholism, morbid obesity and poorly controlled diabetes mellitus.3
Subsequent evaluation
Dexamethasone-corticotropin-releasing hormone test
The dexamethasone-CRH test is useful in patients with ambiguous 24-hour UFC results. The test is done by administering dexamethasone as per the 48-hour 2 mg/d longer low-dose DST, and then CRH (1 µg/kg) is administered intravenously two hours after the last dose of dexamethasone. Cortisol is measured 15 minutes later. If the plasma cortisol value is >1.4 µg/dL, then the patient is likely to have CS.3 To exclude false-positive results, dexamethasone levels should also be measured at the time of CRH administration.3
Midnight serum cortisol test
The midnight serum cortisol test is useful in patients whom, despite a high clinical index of suspicion of CS, had normal 24-hour UFC and full suppression on dexamethasone testing. A sleeping midnight serum cortisol value >1.8 µg/dL or an awake value >7.5 µg/dL increases the probability of CS. The sleeping midnight cortisol test may not be feasible in outpatient settings. It usually requires inpatient admission for a period of 48 hours or longer to avoid false-positive responses due to the stress of hospitalisation.3
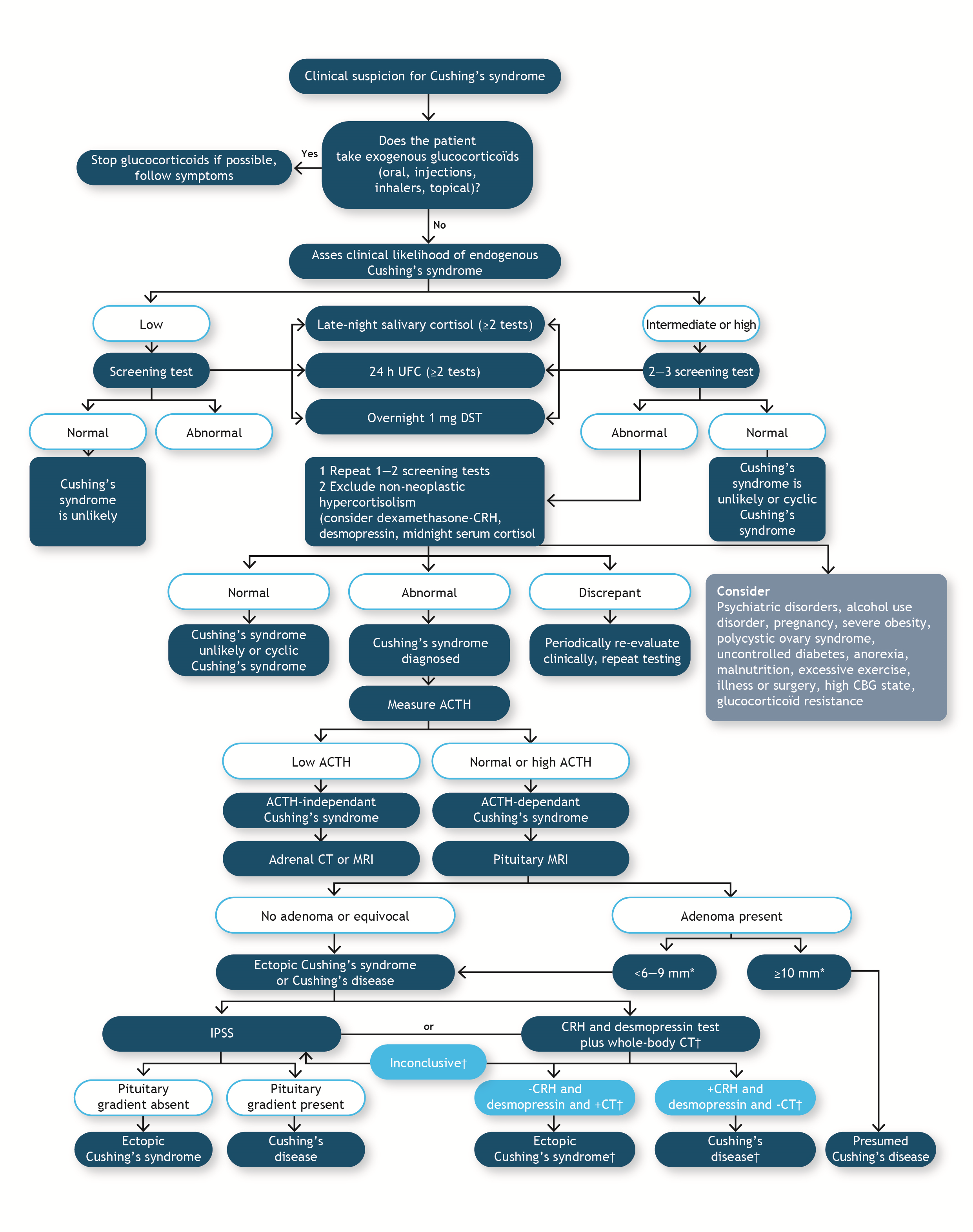 Figure 4. Diagnostic tests to support the diagnosis of Cushing’s syndrome.1ACTH: adrenocorticotrophic hormone; CBG: corticosteroid-binding globulin; CRH: corticotropin-releasing hormone; CT: computed tomography; DST: dexamethasone suppression test; IPSS: inferior petrosal sinus sampling; MRI: magnetic resonance imaging; UFC: urinary free cortisol.
Figure 4. Diagnostic tests to support the diagnosis of Cushing’s syndrome.1ACTH: adrenocorticotrophic hormone; CBG: corticosteroid-binding globulin; CRH: corticotropin-releasing hormone; CT: computed tomography; DST: dexamethasone suppression test; IPSS: inferior petrosal sinus sampling; MRI: magnetic resonance imaging; UFC: urinary free cortisol.Differential diagnosis
Once the diagnosis of CS has been confirmed, the cause of hypercortisolism must be identified. A combination of blood tests and imaging modalities (e.g., magnetic resonance imaging [MRI]/computed tomography [CT] scans) can be employed to determine the cause of CS and localisation of the tumour (Figure 4).1
Plasma adrenocorticotrophic hormone levels
The first step is a blood test in the morning to measure the plasma ACTH level to determine whether CS is ACTH-dependent or -independent. Values <5 pg/mL (1.1 pmol/L) suggest ACTH-independent CS, while an inappropriately normal or elevated ACTH level (>20 pg/mL, 4.4 pmol/L) is consistent with ACTH-dependent CS.6 Levels between 10 and 20 pg/mL are indeterminate, and measurement should be repeated several times. When still uncertain, a CRH test should be performed. If the values are between 1.1 and 4.4 pmol/L, further evaluation with a CRH stimulation test is required to distinguish between pituitary and ectopic sources of ACTH. However, extremely high ACTH levels (>500 pg/mL, 110 pmol/L) indicate an ectopic source of ACTH.5, 6
Corticotropin-releasing hormone stimulation test
The CRH stimulation test is performed by intravenous injection of 1 µg/kg or 100 µg of recombinant ovine or human CRH to stimulate corticotroph tumours to secrete ACTH. In ACTH-independent and ectopic-ACTH CS, there is usually very little or no cortisol or ACTH response to CRH, whereas the response is exaggerated in patients with Cushing’s disease (CD; pituitary-dependent CS). There is generally a >34% increase in ACTH levels and/or >20% increase in cortisol levels within 45 minutes of ovine CRH administration in patients with CD (sensitivity 93%), whereas with human CRH administration, these patients have ≥14% increase in cortisol levels (sensitivity 85%, specificity 100%). However, 7–14% of patients with CD do not respond to this test.6
Desmopressin test
High-dose dexamethasone test
The high-dose DST can be used to distinguish between pituitary and ectopic tumours. Dexamethasone (either 2-mg every 6 hours for 48 hours, or 8-mg overnight) is administered and plasma cortisol levels are evaluated. The principle is that high doses of dexamethasone will suppress cortisol production in pituitary ACTH-secreting tumours, as these tumours retain some sensitivity to glucocorticoid negative feedback but will not suppress an ectopic source of ACTH because of the loss of glucocorticoid negative-feedback inhibition. The high-dose DST lacks optimal diagnostic accuracy, so many endocrinologists do not recommend it unless bilateral inferior petrosal sinus sampling (BIPSS) is not available.6
Tumour localisation
Computed tomography/magnetic resonance imaging
When an adrenal source is suspected in patients with low ACTH levels or when an ectopic ACTH source is suspected, CT/MRI scanning of the adrenals or the whole body, respectively, can be used to localise and identify the lesion responsible for CS.5, 6
Pituitary magnetic resonance imaging
Bilateral inferior petrosal sinus sampling
BIPPS is an invasive procedure and can be used to distinguish between pituitary and non-pituitary sources of CS. Blood samples are taken from the inferior petrosal sinuses that drain the pituitary gland and from a peripheral vein. ACTH levels at both sites are measured before and after CRH stimulation and compared. Patients with an ectopic source of ACTH do not have a gradient between petrosal sinus samples and peripheral ACTH values, whereas a central-to-peripheral ACTH gradient of ≥2 at baseline and/or ≥3 after CRH stimulation is confirmatory of a pituitary tumour.6 This test has sensitivity and specificity of ~95%.5
Octreotide scan
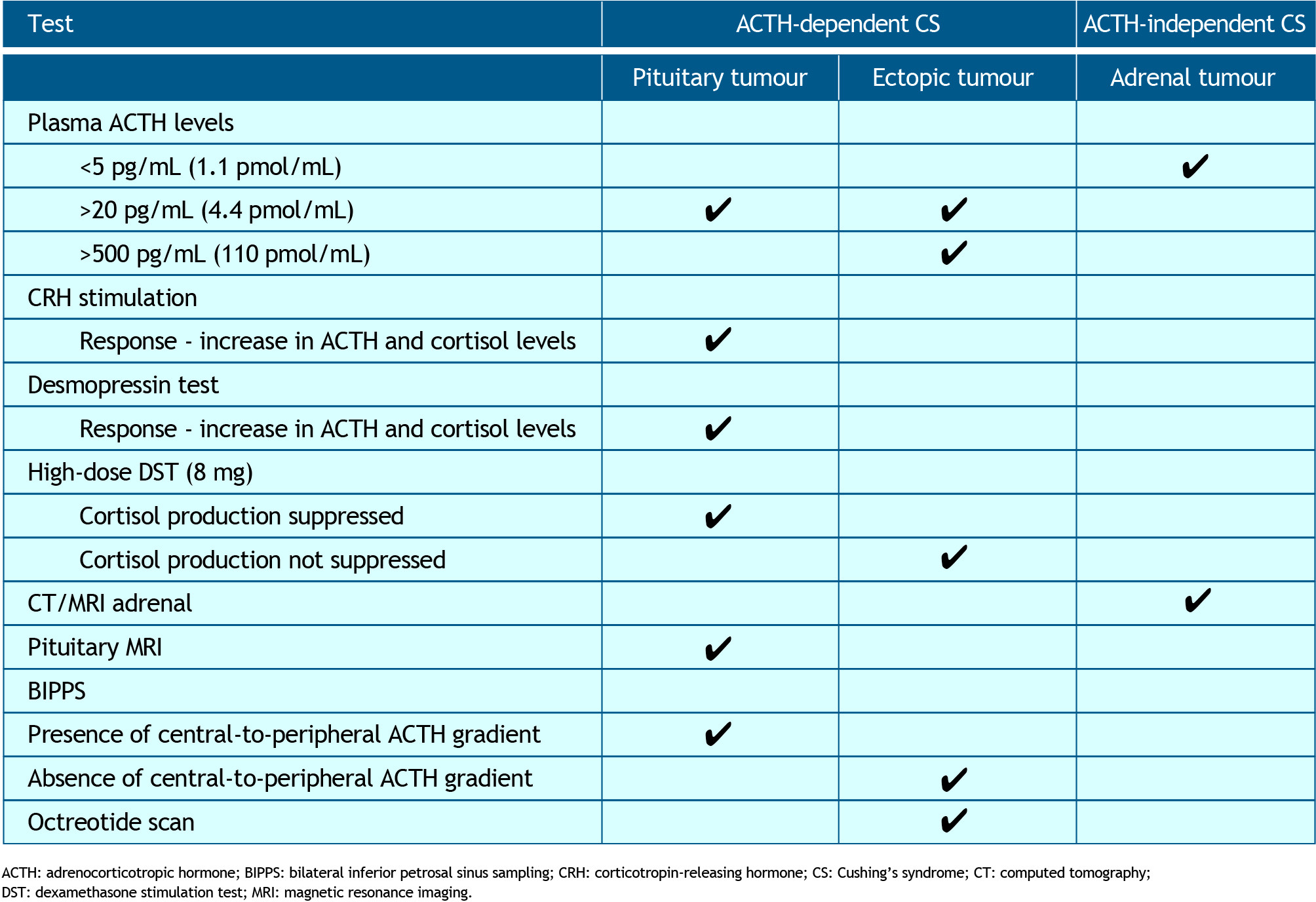 Table 1. Overview of diagnostic tests for distinguishing between causes of CS. The cause of CS identified with each test is shown by ✓ACTH: adrenocorticotropic hormone; BIPPS: bilateral inferior petrosal sinus sampling; CRH: corticotropin-releasing hormone; CS: Cushing’s syndrome; CT: computed tomography; DST: dexamethasone stimulation test; MRI: magnetic resonance imaging.
Table 1. Overview of diagnostic tests for distinguishing between causes of CS. The cause of CS identified with each test is shown by ✓ACTH: adrenocorticotropic hormone; BIPPS: bilateral inferior petrosal sinus sampling; CRH: corticotropin-releasing hormone; CS: Cushing’s syndrome; CT: computed tomography; DST: dexamethasone stimulation test; MRI: magnetic resonance imaging.Diagnostic procedures in suspected adrenocortical carcinoma
Patients with suspected adrenocortical carcinoma (ACC) may also undergo additional tests to identify excess sex steroids and steroid precursors, mineralocorticoids and glucocorticoids. Table 2 provides a summary of the tests recommended by the European Society of Endocrinology in collaboration with the European Network for the Study of Adrenal Tumors in patients with suspected ACC.10
 Table 2. Diagnostic work-up in patients with suspected or proven adrenocortical carcinoma. Reproduced from Fassnacht et al., 2018,10 with permission under a Creative Commons Attribution 4.0 International License.ACTH: adrenocorticotropic hormone; DHEA-S: dehydroepiandrosterone sulphate; CT: computed tomography; FDG-PET: 18F-fluorodeoxyglucose positron emission tomography; MRI: magnetic resonance imaging.
Table 2. Diagnostic work-up in patients with suspected or proven adrenocortical carcinoma. Reproduced from Fassnacht et al., 2018,10 with permission under a Creative Commons Attribution 4.0 International License.ACTH: adrenocorticotropic hormone; DHEA-S: dehydroepiandrosterone sulphate; CT: computed tomography; FDG-PET: 18F-fluorodeoxyglucose positron emission tomography; MRI: magnetic resonance imaging.References
- Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol. 2021; 9: 847-875. https://pubmed.ncbi.nlm.nih.gov/34687601.
- Gilbert R, Lim EM. The diagnosis of Cushing’s syndrome: an endocrine society clinical practice guideline. Clin Biochem Rev. 2008; 29: 103-106. https://pubmed.ncbi.nlm.nih.gov/19107223.
- Nieman LK, Biller BMK, Findling JW, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008; 93: 1526-1540. https://pubmed.ncbi.nlm.nih.gov/18334580.
- Ceccato F, Boscaro M. Cushing’s syndrome: screening and diagnosis. High Blood Press Cardiovasc Prev. 2016; 23: 209-215. https://pubmed.ncbi.nlm.nih.gov/27160717.
- Pappachan JM, Hariman C, Edavalath M, Waldron J, Hanna FW. Cushing’s syndrome: a practical approach to diagnosis and differential diagnoses. J Clin Pathol. 2017; 70: 350-359. https://pubmed.ncbi.nlm.nih.gov/28069628.
- Sharma ST, Nieman LK, Feelders RA. Cushing’s syndrome: epidemiology and developments in disease management. Clin Epidemiol 2015; 7: 281-293. https://pubmed.ncbi.nlm.nih.gov/25945066.
- Frete C, Corcuff JB, Kuhn E, et al. Non-invasive diagnostic strategy in ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 2020; 105. https://pubmed.ncbi.nlm.nih.gov/32594169.
- Ioachimescu AG, Remer EM, Hamrahian AH. Adrenal incidentalomas: a disease of modern technology offering opportunities for improved patient care. Endocrinol Metab Clin North Am. 2015; 44: 335-354. https://pubmed.ncbi.nlm.nih.gov/26038204.
- Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab. 2003; 88: 5593-5602. https://pubmed.ncbi.nlm.nih.gov/14671138.
- Fassnacht M, Dekkers OM, Else T, et al. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol. 2018; 179: G1-g46. https://pubmed.ncbi.nlm.nih.gov/30299884.
Disease management
Patients with Cushing’s syndrome (CS) have increased morbidity and mortality rates compared with the general population. Providing effective and timely management is therefore essential. Here, we discuss treatment goals, the different comorbidities, current guidelines and the various lines of treatment for CS, including transsphenoidal surgery, radiotherapy, bilateral adrenalectomy, pharmacological interventions and future treatments.
Clinical treatment guidelines
Managing comorbidities
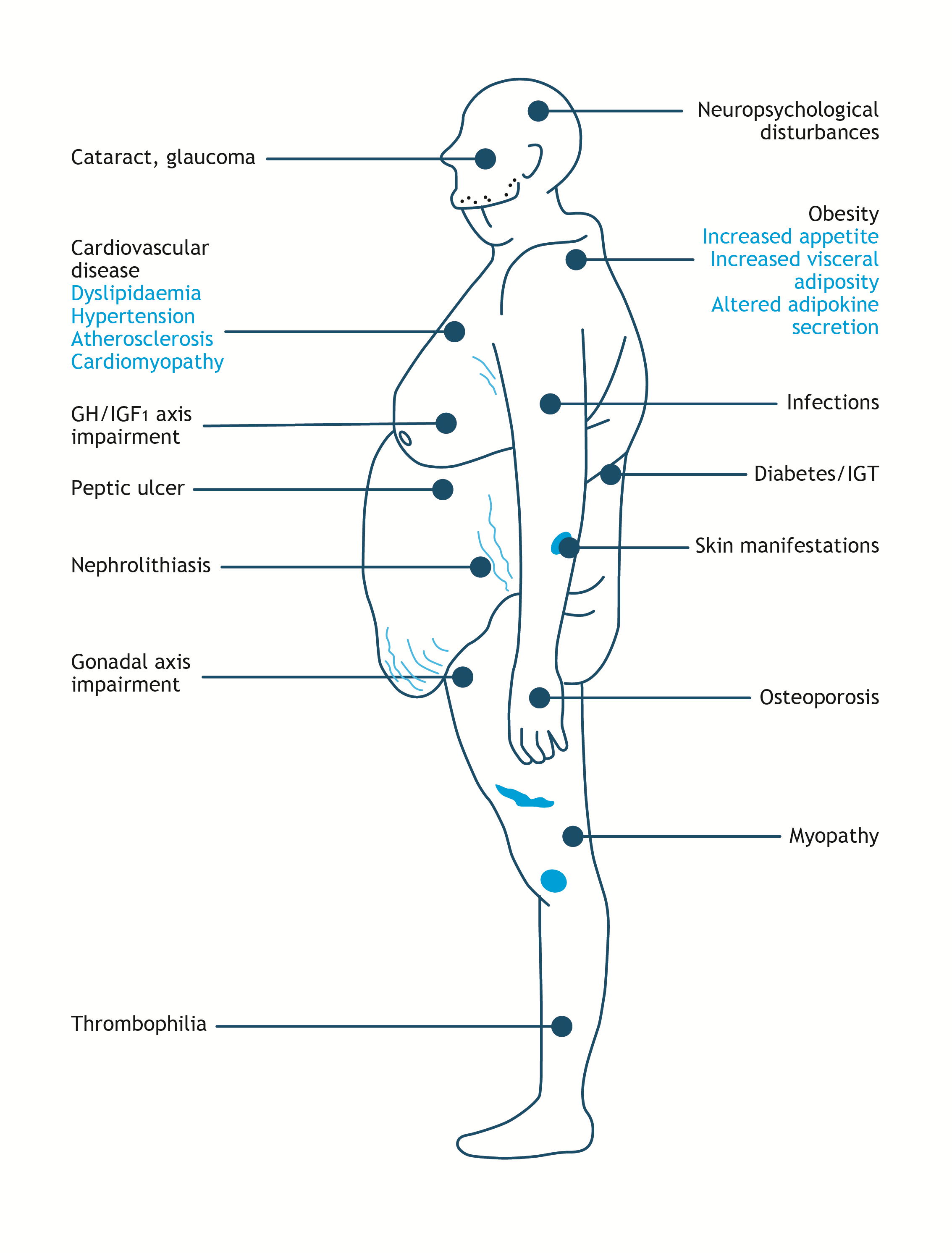 Figure 5. Comorbidities in Cushing’s Syndrome. Adapted from Arnaldi et al., 2003 and Ferraù and Korbonits, 2015.5, 6GH/IGF1: growth hormone/insulin-like growth factor 1; IGT: impaired glucose tolerance.
Figure 5. Comorbidities in Cushing’s Syndrome. Adapted from Arnaldi et al., 2003 and Ferraù and Korbonits, 2015.5, 6GH/IGF1: growth hormone/insulin-like growth factor 1; IGT: impaired glucose tolerance.Treatment
CS treatment aims to rapidly normalise the cortisol levels, and consequently, to improve the metabolic and biochemical abnormalities associated with the disorder. Current guidelines provide advice on the surgical, radiological and medical management of CS with clear recommendations on lines of therapy and patient suitability.1, 2
First-line treatment
 Figure 6. Post-surgical treatment is based on subsequent serum cortisol levels.1DST: dexamethasone suppression test; HPA: hypothalamic-pituitary-adrenal; LNSC: late-night salivary cortisol.
Figure 6. Post-surgical treatment is based on subsequent serum cortisol levels.1DST: dexamethasone suppression test; HPA: hypothalamic-pituitary-adrenal; LNSC: late-night salivary cortisol.Second-line treatment
Both the Pituitary Society and the Endocrine Society suggest a shared decision-making approach to allow patients a choice in their treatment when surgery is not possible or is unsuccessful.1, 2 The factors that should be taken into account when developing a treatment plan are presented in Figure 7.
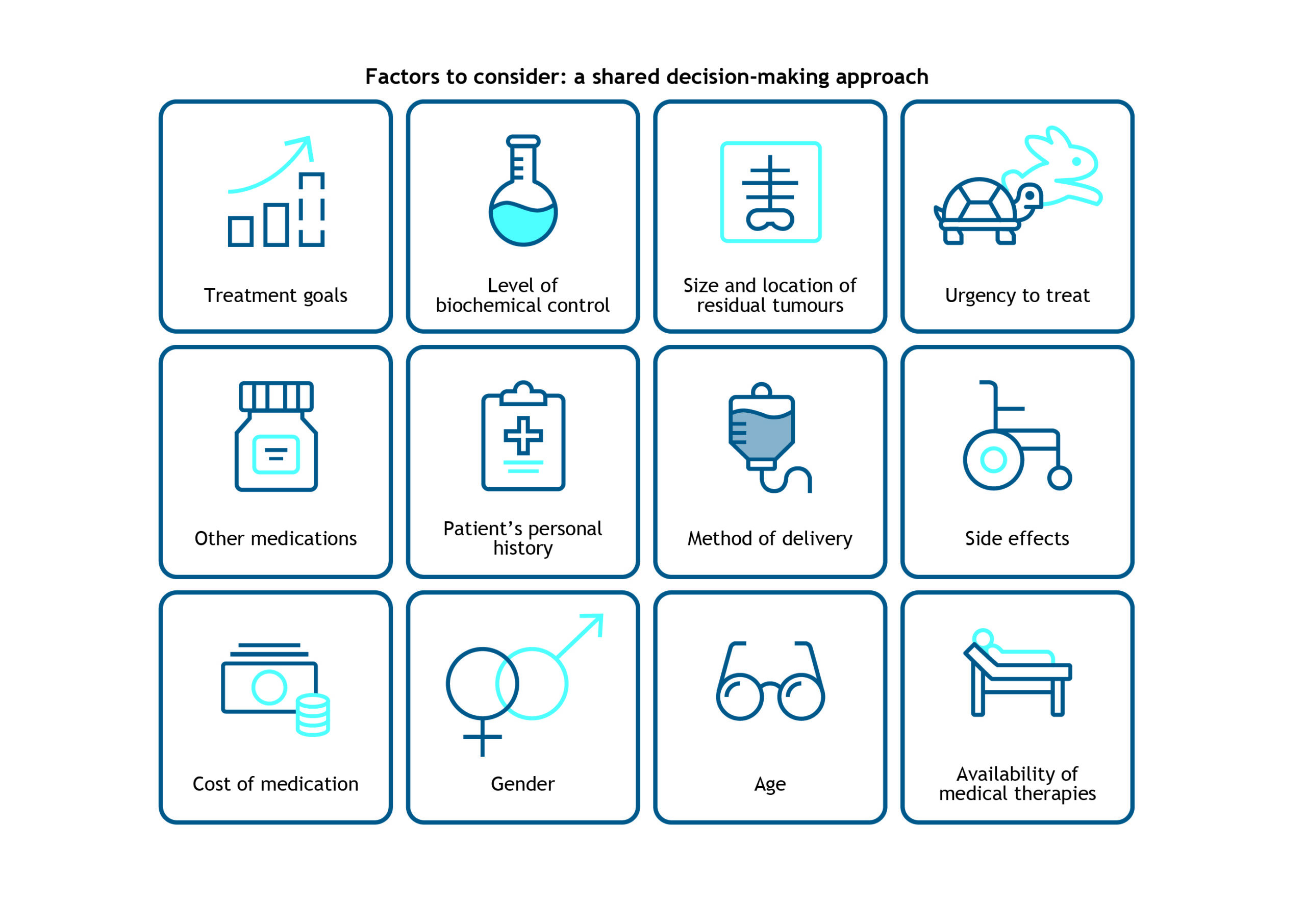 Figure 7. Factors to consider when developing a second-line treatment plan.1
Figure 7. Factors to consider when developing a second-line treatment plan.1Possibilities in Cushing’s disease
Repeat transsphenoidal surgery
TSS should be repeated when there is evidence of incomplete resection of the pituitary adenoma or imaging shows a pituitary lesion, and especially if the first surgery was not performed by an expert team.1
Radiation therapy/radiosurgery
Radiation therapy/radiosurgery is suitable in patients with persistent or recurrent disease and when the tumour is visible in the magnetic resonance imaging (MRI), especially, when surgery was performed despite the invasiveness of the tumour. Because the effects of radiation occur over a long time, it is important to normalise cortisol levels through medical therapy while waiting for the radiation to take effect. Radiation is delivered as either external beam therapy or as stereotactic surgery.1, 2 Radiation therapy can be very effective, but its effects can take as long as ten years to manifest, and it can lead to long-term hypopituitarism. Stereotactic radiosurgery is associated with more rapid effectiveness, but has a relapse rate of 20%.2, 7
Bilateral adrenalectomy
Laparoscopic bilateral adrenalectomy is an alternative secondary approach for patients with CD unsuccessfully treated by pituitary surgery. It offers immediate control of hypercortisolism, but is generally only indicated in patients for whom medical therapy has failed or is not suitable. Additionally, as the surgery results in the permanent removal of the adrenal gland, the patient will require life-long treatment with glucocorticoid and mineralocorticoid replacement therapies.1, 7 There is also the risk of developing Nelson’s syndrome (continued growth of the pituitary releasing ACTH), so close endocrinological follow-up with ACTH evaluation and regular MRI scans are mandatory in patients whom undergo bilateral adrenalectomy. Nevertheless, this will immediately control the cortisol excess, and should be considered as a reasonable option, especially in the absence of an obvious pituitary tumour.1, 7, 10
Medical therapies
 Figure 8. Medical treatments and suitability for patients with Cushing’s syndrome.ACTH: adrenocorticotrophic hormone; AVP: arginine vasopressin; CD: Cushing’s disease; CRH: corticotropin-releasing hormone; DAs: dopamine receptor agonists; GR: glucocorticoid receptor, GRE: glucocorticoid- response elements; SRLs: somatostatin receptor ligands; TSS: transsphenoidal surgery.
Figure 8. Medical treatments and suitability for patients with Cushing’s syndrome.ACTH: adrenocorticotrophic hormone; AVP: arginine vasopressin; CD: Cushing’s disease; CRH: corticotropin-releasing hormone; DAs: dopamine receptor agonists; GR: glucocorticoid receptor, GRE: glucocorticoid- response elements; SRLs: somatostatin receptor ligands; TSS: transsphenoidal surgery.Neuromodulators of adrenocorticotrophic hormone release
Guidelines recommend pituitary-directed therapies in patients with CD in whom surgery is contraindicated or whom have persistent disease after TSS. These therapies directly target the corticotroph pituitary adenomas to reduce ACTH hypersecretion. Two currently available classes of medications that act centrally are dopamine type 2 receptor agonists (DAs), and somatostatin receptor ligands (SRLs).1, 2
Cabergoline
Cabergoline is a DA indicated for the inhibition of lactation and treatment of hyperprolactinaemia disorders. It acts by directly stimulating D2-dopamine receptors on pituitary lactotrophs, thus inhibiting prolactin secretion. Cabergoline for CS is typically initiated at 0.5 mg per week, with titration to a maximal dose of 7 mg/week. It has been shown to normalise urinary free cortisol (UFC) levels in 30–40% of the patients,1, 7 improve weight, glucose levels and hypertension in 25–40% of complete responders and shrink tumours in 50% of patients.12 In a study of 30 CS patients, long-term treatment resulted in complete remission in 30% of patients, although some patients subsequently relapsed.13 In one of the largest cohort studies in patients with CD, cabergoline had acceptable tolerability and provided long-term control of hypercortisolism at relatively low doses. Among the 53 patients receiving low-dose cabergoline monotherapy (1.5 mg/week), 21 patients (40%) achieved normal UFC values and clinical improvement within 12 months of treatment, which was sustained through 32.5 months in 12 patients (23%). Among patients receiving cabergoline add-on therapy (1.0 mg/week) for 19 months, hypercortisolism was controlled in 56% of patients during the first year of treatment.14 Adverse effects with cabergoline may include headache, dizziness, gastrointestinal discomfort, asthenia and cardiac valve fibrosis at high doses.1, 7
Pasireotide
Pasireotide is a somatostatin receptor ligand indicated for the treatment of CD. Pasireotide binds with high affinity to somatostatin receptors 1–3 and 5, i.e. four of the five human somatostatin receptors, inhibiting tumour growth and lowering ACTH production.1 A long-acting monthly formulation has been recently trialled, with slightly higher response rates compared with the subcutaneous formulation. Nevertheless, only patients with relatively mild disease responded, and the issue of hyperglycaemia and frank diabetes remains problematic.15 In a phase III trial of 150 adults with CD, long-acting pasireotide (10 mg or 30 mg), administered intramuscularly, normalized UFC in about 40% of patients after 7 months.16 Moreover, the safety profile of long-acting pasireotide was similar to that of subcutaneous pasireotide (600–900 µg twice daily).17 Pasireotide should be given for as long as clinical benefit is observed, but blood sugar levels must be carefully monitored due to an increased risk of hyperglycaemia. Other potential adverse effects are diarrhoea, nausea, cholelithiasis, transient increases in liver function tests and hypothyroidism.1, 2
Adrenal steroidogenesis inhibitors
Ketoconazole
Ketoconazole is an antifungal agent approved by the European Medicines Agency for the treatment of CS and has a rapid onset of action. It is an imidazole derivative that inhibits steroid synthesis through inhibition of cytochrome P450 enzymes; 17, 20-lyase; 11β-hydroxylase; 17-hydroxylase and side-chain cleavage enzymes. These effects are dose dependent and completely and rapidly reversible upon drug discontinuation. The recommended dose of ketoconazole is 400–1600 mg (the usual maximal dose being 1200 mg/day) (every 6–8 hours dosing). In a large retrospective study, ketoconazole was associated with a normalization of UFC in 50% of patients and at least a 50% reduction in UFC in 25% of patients.18 Ketoconazole requires an acidic environment to be most effective, therefore, it has reduced efficacy if used with proton pump inhibitors. Potential adverse effects include serious hepatotoxicity (requiring liver function test monitoring at treatment initiation and after each dose adjustment), gastrointestinal discomfort, decreased testosterone levels, gynaecomastia and adrenal insufficiency.7 Since ketoconazole can potentially interact with a number of drugs, clinicians should be aware of their patients’ medication list to avoid problematic interactions.2
Metyrapone
Mitotane
Mitotane inhibits several steroidogenic enzymes and has a slow-onset, long-lasting adrenolytic action in steroid-secreting adrenocortical cells. It suppresses hypercortisolism in 80% of cases. Mitotane is indicated for the symptomatic treatment of advanced (unresectable, metastatic or relapsed) ACC,1, 2 but it has also been delivered in patients with CD.22 European Society for Medical Oncology guidelines recommend a starting dose of 1.5 g/day, increasing within 4–6 days up to 6 g/day. Blood levels should be monitored and a target level of 14–20 mg/L should be sought for adrenal carcinoma (whereas target level of mitotane is 10 mg/L for CD); approximately 50% of patients achieve this target within 3 months.23 Mitotane increases cortisol-binding globulin levels, with corresponding increases in plasma cortisol; therefore, biochemical monitoring should be done with UFC or salivary cortisol measurements.1 Moreover, mitotane usually induces adrenal insufficiency requiring higher doses of hydrocortisone than in classical adrenal insufficiency (up to 40–50 mg/day because of the enzymatic inducer effect of the drug).23 It is teratogenic and not well tolerated; adverse effects may include hepatotoxicity, gastrointestinal discomfort, hypercholesterolaemia, gynaecomastia, prolonged bleeding time, dizziness, ataxia, dysarthria, memory loss and adrenal insufficiency.7
Osilodrostat
Osilodrostat is an 11β-hydroxylase and aldosterone synthase inhibitor that effectively reduces cortisol levels with a high success rate. It is approved in the US for CS patients whom have failed surgery or are contraindicated for surgery.24 It is well-tolerated and associated with improvements in bodyweight, psychiatric disturbances, blood pressure, total cholesterol, low-density lipoprotein cholesterol, fasting serum glucose and glycated haemoglobin concentrations. The most common adverse effects are nausea, anaemia, headache and adverse events related to hypocortisolism.2
Etomidate
Etomidate is an intravenous medication originally developed as an anaesthetic, but which rapidly normalises cortisol levels by inhibiting 11β-hydroxylase and cholesterol side-chain cleavage.2 Guidelines indicate that etomidate can be used to manage hypercortisolaemia in severely ill patients of any age whom cannot receive surgery immediately or take oral medications. It has a quick onset of action but requires monitoring in a high-dependency unit. First, a loading dose of 3–5 mg is administered followed by a maintenance dose of 0.03–0.1 mg/kg/h (2.5–3.0 mg/h) to achieve stable serum cortisol levels of 10–20 µg/dL (280–560 nmol/L). Etomidate is not sedative at these doses, but dose reduction might be needed if renal failure occurs.1
Levoketoconazole
Levoketoconazole is an enantiomer of ketoconazole. Both the US Food and Drug Administration and European Medicines Agency have granted orphan drug status to levoketoconazole for treatment of endogenous CS. The recommended dose is 300–1200 mg, given orally twice a day.2 Levoketoconazole has been shown to normalise UFC levels in 31% of patients,25 with a lower rate of withdrawal compared with placebo (41% vs 96%).26 The most common adverse effects include gastrointestinal disturbances, headache, oedema, increased liver enzymes and adrenal insufficiency. There is a possibility of interaction with other drugs.2
Glucocorticoid receptor blockers - Mifepristone
Future treatments
Specific management of adrenal carcinomas
The initial management of adrenal carcinomas involves resection of the lesion, unless surgery is not possible. Since patients with ACC have a poor prognosis, complete resection should be the goal of surgery.1 Patients with ACC should undergo evaluation of tumour size and stage of development and treated accordingly. Follow-up care should be provided by an adrenal cancer-specific multidisciplinary team, and may include cytotoxic chemotherapy and adjuvant radiotherapy or medical therapy (mitotane) to achieve eucortisolism.1 Severe hypercortisolism can also be managed with a combination of steroidogenesis inhibitors.20, 21
References
- Nieman LK, Biller BM, Findling JW, et al. Treatment of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2015; 100: 2807-2831. https://pubmed.ncbi.nlm.nih.gov/26222757.
- Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol. 2021; 9: 847-875. https://pubmed.ncbi.nlm.nih.gov/34687601.
- Clayton RN, Jones PW, Reulen RC, et al. Mortality in patients with Cushing’s disease more than 10 years after remission: a multicentre, multinational, retrospective cohort study. Lancet Diabetes Endocrinol. 2016; 4: 569-576. https://pubmed.ncbi.nlm.nih.gov/27265184.
- Ferriere A, Tabarin A. Cushing’s syndrome: treatment and new therapeutic approaches. Best Pract Res Clin Endocrinol Metab. 2020; 34: 101381. https://pubmed.ncbi.nlm.nih.gov/32035797.
- Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab. 2003; 88: 5593-5602. https://pubmed.ncbi.nlm.nih.gov/14671138.
- Ferraù F, Korbonits M. Metabolic comorbidities in Cushing’s syndrome. Eur J Endocrinol. 2015; 173: M133-157. https://pubmed.ncbi.nlm.nih.gov/26060052.
- Sharma ST, Nieman LK, Feelders RA. Cushing’s syndrome: epidemiology and developments in disease management. Clin Epidemiol 2015; 7: 281-293. https://pubmed.ncbi.nlm.nih.gov/25945066.
- Hinojosa-Amaya JM, Varlamov EV, McCartney S, Fleseriu M. Hypercortisolemia recurrence in Cushing’s disease: A diagnostic challenge. Front Endocrinol. 2019; 10: 740-740. https://pubmed.ncbi.nlm.nih.gov/31787930.
- Rizk A, Honegger J, Milian M, Psaras T. Treatment options in Cushing’s disease. Clin Med Insights Oncol. 2012; 6: 75-84. https://pubmed.ncbi.nlm.nih.gov/22346367.
- Reincke M, Albani A, Assie G, et al. Corticotroph tumor progression after bilateral adrenalectomy (Nelson’s syndrome): systematic review and expert consensus recommendations. Eur J Endocrinol. 2021; 184: 1-16. https://pubmed.ncbi.nlm.nih.gov/33444221.
- Pivonello R, De Leo M, Cozzolino A, Colao A. The treatment of Cushing’s disease. Endocr Rev. 2015; 36: 385-486. https://pubmed.ncbi.nlm.nih.gov/26067718.
- Pivonello R, De Martino MC, Cappabianca P, et al. The medical treatment of Cushing’s disease: effectiveness of chronic treatment with the dopamine agonist cabergoline in patients unsuccessfully treated by surgery. J Clin Endocrinol Metab. 2009; 94: 223-230. https://pubmed.ncbi.nlm.nih.gov/18957500.
- Godbout A, Manavela M, Danilowicz K, Beauregard H, Bruno OD, Lacroix A. Cabergoline monotherapy in the long-term treatment of Cushing’s disease. Eur J Endocrinol. 2010; 163: 709-716. https://pubmed.ncbi.nlm.nih.gov/20702648.
- Ferriere A, Cortet C, Chanson P, et al. Cabergoline for Cushing’s disease: a large retrospective multicenter study. Eur J Endocrinol. 2017; 176: 305-314. https://pubmed.ncbi.nlm.nih.gov/28007845.
- John Newell-Price, Stephan Petersenn, Beverly M K Biller, Michael Roughton, Ravichandran S, Lacroix A. Once-monthly injection of pasireotide LAR reduces urinary free cortisol (UFC) levels in patients with Cushing’s disease: results from a randomised, multicentre, phase III trial. Endocrine Abstracts
- Lacroix A, Gu F, Gallardo W, et al. Efficacy and safety of once-monthly pasireotide in Cushing’s disease: a 12 month clinical trial. Lancet Diabetes Endocrinol. 2018; 6: 17-26. https://pubmed.ncbi.nlm.nih.gov/29032078.
- Colao A, Petersenn S, Newell-Price J, et al. A 12-month phase 3 study of pasireotide in Cushing’s disease. N Engl J Med. 2012; 366: 914-924. https://pubmed.ncbi.nlm.nih.gov/22397653.
- Castinetti F, Guignat L, Giraud P, et al. Ketoconazole in Cushing’s disease: Is it worth a try? J Clin Endocrinol Metab. 2014; 99: 1623-1630. https://pubmed.ncbi.nlm.nih.gov/24471573.
- Al-Salama ZT. Metyrapone in Cushing’s syndrome: a profile of its use. Drugs Ther Perspect. 2021; 37: 393-406. https://doi.org/10.1007/s40267-021-00853-y.
- Corcuff JB, Young J, Masquefa-Giraud P, Chanson P, Baudin E, Tabarin A. Rapid control of severe neoplastic hypercortisolism with metyrapone and ketoconazole. Eur J Endocrinol. 2015; 172: 473-481. https://pubmed.ncbi.nlm.nih.gov/25624013.
- Kamenický P, Droumaguet C, Salenave S, et al. Mitotane, metyrapone, and ketoconazole combination therapy as an alternative to rescue adrenalectomy for severe ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 2011; 96: 2796-2804. https://pubmed.ncbi.nlm.nih.gov/21752886.
- Baudry C, Coste J, Bou Khalil R, et al. Efficiency and tolerance of mitotane in Cushing’s disease in 76 patients from a single center. Eur J Endocrinol. 2012; 167: 473-481. https://pubmed.ncbi.nlm.nih.gov/22815335.
- Berruti A, Baudin E, Gelderblom H, et al. Adrenal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012; 23: vii131-vii138. https://pubmed.ncbi.nlm.nih.gov/22997446.
- S. Food and Drug Administration. (2020). “FDA approves new treatment for adults with Cushing’s disease.” March 17, 2022, from https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-adults-cushings-disease.
- Fleseriu M, Pivonello R, Elenkova A, et al. Efficacy and safety of levoketoconazole in the treatment of endogenous Cushing’s syndrome (SONICS): a phase 3, multicentre, open-label, single-arm trial. The Lancet Diabetes & Endocrinology. 2019; 7: 855-865. https://pubmed.ncbi.nlm.nih.gov/31542384.
- Zacharieva S, Pivonello R, Elenkova A, et al. Safety and efficacy of levoketoconazole in the treatment of endogenous Cushing’s syndrome (LOGICS): results from a double-blind, placebo-controlled, randomized withdrawal study. J Endocr Soc. 2021; 5: A526-A526. https://pubmed.ncbi.nlm.nih.gov/PMC8090707.
- Raff H, Carroll T. Cushing’s syndrome: from physiological principles to diagnosis and clinical care. J Physiol. 2015; 593: 493-506. https://pubmed.ncbi.nlm.nih.gov/25480800.
- Fleseriu M, Biller B, Findling J, Molitch M, Schteingart D, Gross C. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing’s syndrome. J Clin Endocrinol Metab. 2012; 97: 2039-2049. https://pubmed.ncbi.nlm.nih.gov/22466348.
- Cuevas-Ramos D, Lim DST, Fleseriu M. Update on medical treatment for Cushing’s disease. Clin Diabetes Endocrinol. 2016; 2: 16. https://pubmed.ncbi.nlm.nih.gov/28702250.
Resources
EU: Adrenals.eu
A website for patients with adrenal disorders, their families, carers, and the healthcare practitioners they engage with, to work towards better diagnosis and care. This website has editions in Dutch, German and Danish.
France: Association Surrénales
The Association Surrénales is an organisation for sufferers of adrenal disease which aims to provide support, raise awareness and promote scientific research. The website is in French.
Germany: Netzwerk Glandula
Netzwerk Glandula is a network forum for exchange between doctors and patients with pituitary and adrenal diseases. This website is in German.
Spain: Ayuda Pacientes Con Enfermedad Hipofisaria
UK: Association for Multiple Endocrine Neoplasia Disorders (AMEND)
UK: The Pituitary Foundation
UK: ACC Support UK
USA: Cushing’s Support and Research Foundation